Pulmoner Arteriyel Hipertansiyon (PAH)
Pulmoner hipertansiyon; vücutta dolaşan kanı oksijenlenmek üzere kalpten akciğerlere getiren damarlarda (pulmoner arterlerde) kan basıncının artmasıdır. Pulmoner hipertansiyona yol açabilecek onlarca hastalık mevcuttur. Bunların içerisindeki bir grup olan Pulmoner Arteriyel Hipertansiyon; hızlı ilerleyerek, yaşamı tehdit edebilir¹.
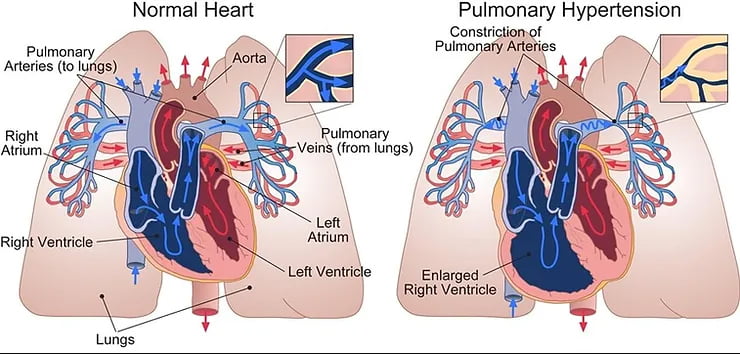
Normal şartlar altında kalbin sağ tarafı (sağ atriyum ve sağ ventrikül), kirli kanı temizlenmek (oksijenlenmek) üzere akciğerlere pulmoner arterler yoluyla gönderir. Pulmoner hipertansiyon geliştiğinde, pulmoner arterler daralır. Daralan pulmoner arterlere kan pompalamaya çalışan zorlanmaya başlar ve kalbin sağ tarafı genişler¹.
Pulmoner hipertansiyon kalbin sağ tarafı ve akciğer arterlerini etkileyen bir tür kan basıncı yüksekliğidir².
Artan basınç, kalbin sağ ventrikülün, kanı akciğerlere pompalamak için daha fazla güç harcamasını, bu durum da zamanla kalp kasında güçsüzlük ve yetmezliğe yol açar².
Normal pulmoner arter basıncı, dinlenme halinde 14 ± 3 mmHg, üst normal limiti ise 20 mmHg’dır³.
Pulmoner hipertansiyonda ise ortalama pulmoner arter basıncı, PAB⩾25 mmHg’dır³.
Pulmoner arteriyel hipertansiyon (PAH), akciğer hastalıkları, KTEPH ya da nadir hastalıklar gibi diğer prekapiller PH nedenleri olmaksızın, pulmoner arter uç basıncı PAWB ≤15 mmHg ve PVD >3 Wood ünitesi (WU) ile tanımlanan bir grup PH hastasını ifade etmektedir³.
EPİDEMİYOLOJİ
Literatürde PH insidansına yönelik veri yetersizdir³.
Çeşitli kayıt çalışmalarında PAH’nin epidemiyolojisi tanımlanmaktadır. Erişkin nüfusta hesaplanan en düşük PAH ve idiyopatik PAH (İPAH) prevalans tahmini, sırasıyla, 15/1 milyon ve 5,9/1 milyon olgudur³.
En düşük PAH insidans tahmini ise erişkin nüfusta yıl başına 2,4/1 milyon olgudur³.
Avrupa’da, PAH prevalansı ve insidansı, sırasıyla, 15-60/ 1 milyon hasta ve yıl başına 5-10/1 milyon olgu aralığındadır³.
SINIFLANDIRMA²
Grup 1: Pulmoner arteriyel hipertansiyon
Grup 2: Sol kalp hastalığı kaynaklı Pulmoner hipertansiyon
Grup 3: Akciğer hastalığı ve/veya hipoksi kaynaklı Pulmoner hipertansiyon
Grup 4: Kronik pulmoner emboli kaynaklı Pulmoner hipertansiyon
Grup 5: Nedeni bilinmeyen/multifaktöriyel Pulmoner hipertansiyon
BELİRTİLER²
Hastalığın erken aşamalarında semptomlar nonspesifik olduğu için hastalığı tespit etmek zordur.
Nefes darlığı (başlangıçta egzersiz yaparken ve sonrasında dinlenme sırasında)
Yorgunluk
Baş dönmesi veya bayılma nöbetleri (senkop)
Göğüs de basınç hissi veya göğüs ağrısı
Ayak bileklerinde, bacaklarında ve nihayetinde karında şişlik (ödem)
Dudaklar ve ciltte mavimsi renk (siyanoz)
Çarpıntı

PH tanısı, semptomlara ve fizik muayeneye dayalı klinik kuşku gerektirir. Bunun için bir dizi incelemenin yapılması gerekir. Bunlardan birkaçı aşağıda sıralanmıştır:
EKG (Elektrokardiyografi)
Akciğer grafisi
Solunum fonksiyon testleri ve arter kan gazları
Ekokardiyografi
Ventilasyon- perfüzyon sintigrafisi
Yüksek çözünürlüklü BT
Kontrastlı BT ve pulmoner anjiyografi
Kardiyak MR
Sağ kalp kateterizasyonu (SKK) ve vazoreaktivite
Genetik testler
TEDAVİ
Pulmoner hipertansiyon tanı ve tedavisi, neden olan hastalığın PAH sınıflamadaki yerine göre değişir. Birçok uzmanlık alanını ilgilendiren bir hastalık olması nedeniyle PAH hastaları multidisipliner yaklaşımla incelenmeli, tedavi ve izlemleri PAH merkezlerindeki deneyimli uzmanlarca yapılmalıdır⁴.
PAH hastalarında genel tedavi hedefi³;
Egzersiz kapasitesini, yaşam kalitesini, sağ ventrikül fonksiyonunu iyileştirerek, mortaliteyi azaltarak hastayı düşük risk grubuna sokulması,
Hastanın mümkün olduğunda WHO-FC II’ye getirilmesi ve/veya burada tutulması,
6DYM’nin normal ya da normale yakın hale getirilmesidir. – 6DYM >440 m’lik bir eşik değer benimsenmektedir.
PAH hastaları için mevcut tedavi stratejisi üç ana adıma ayrılabilir³:
Başlangıç yaklaşımı, genel önlemleri, destek tedaviyi (oral antikoagülanlar, diüretikler, O2, digoksin), uzmanlaşmış merkezlere sevki ve kronik KKB tedavisi endikasyonu için akut vazoreaktivite testini içerir.
İkinci adım, başlangıç tedavisi olarak vazoreaktif hastalarda yüksek doz KKB’yi, vazoreaktif olmayan hastalarda ise PAH için onaylanmış ilaçları, hastanın prognostik riskine göre ve her bir ürün ya da ürünlerin kombinasyonları için var olan öneri derecesine ve kanıt düzeyine göre vermektir.
Üçüncü adım, başlangıç tedavi stratejisine alınan yanıtla ilgilidir; yanıt yetersizse, onaylı ilaç kombinasyonlarının kullanılması ve akciğer transplantasyonu önerilir.
PAH tedavisi ile hastalığın klinik kötüleşme hızı yavaşlatılabilmekte ve sağ kalım kısmen uzatılabilmektedir. Maalesef PAH, halen, tamamen tedavi edilir değil, kontrol edilebilir bir hastalıktır. PAH tedavisinin hedefleri, hastanın fonksiyonel evresini düzeltmek, klinik kötüleşmeyi durdurmak ya da iyileştirmek, yaşam kalitesini düzeltmek ve sağ kalım süresini uzatmaktır⁴.
PAH fizyopatolojisine yönelik yeni ilaçlar geliştirilmiştir. Bunlar endotel reseptör antagonistleri (oral bosentan, ambrisentan, sitaksentan); prostanoidler (inhaler iloprost, subkutan/iv treprostinil ve iv epoprostenol) ve fosfodiesteraz inhibitörleridir (oral sildenafil, tadalafil, vardanafil). Bu ilaçlarla yapılmış randomize kontrollü çalışmalarla, ilaçların her fonksiyonel evredeki etki ve güvenliği konusunda kanıtlar ve kanıt düzeyleri tanımlanmıştır. Tedavi kılavuzlarında her fonksiyonel evredeki hastaya önerilen ilaçlar, kanıt düzeylerine göre gösterilmiştir⁴.
PAH tedavisi başlanan her hasta, 3 ay ara ile tedavinin yanıtı açısından izlenmelidir. Tek ilaç tedavisi ile hedeflenen yanıt elde edilmezse, iki ya da üç ilaç birlikte yapılabilen “kombinasyon” tedavisine geçilmelidir⁴.
İlaç tedavisinin başarısız ya da yetersiz olduğu ve zaman zaman senkop geçiren ileri dönem PAH hastalarında mortaliteyi önlemek amacı ile balon atriyal septostomi (BAS) yada akciğer yada kalp akciğer transplantasyonu uygulanmalıdır⁴.
PAH hastalarının gebe kalması önlenmeli, kalmışlarsa da gebelik sonlandırılmalıdır⁴.

REFERANSLAR
https://www.mayoclinic.org/diseases-conditions/pulmonary-hypertension/symptoms-causes/syc-20350697
2015 ESC/ERS Pulmoner Hipertansiyon Tanı ve Tedavi Kılavuzu
Resim 1: https://www.shutterstock.com/tr/image-vector/3d-vector-human-respiratory-system-lungs- 1511668082
Resim 2: https://www.achaheart.org/your-heart/health-information/pulmonary-hypertension/
Resim 3: https://www.mayoclinic.org/diseases-conditions/pulmonary-hypertension/symptoms-causes/syc-20350697



